Fenilchetonuria
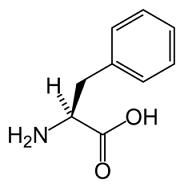
Fenilchetonuria o PKU è un disordine metabolico che causa l'incapacità del corpo di metabolizzare l'aminoacido fenilalanina nel fegato. È una malattia ereditaria causata dalla mancanza di un enzima conosciuto come fenil alanina idrossilasi (abbreviato in FAOH) o tirosina idrossilasi (DHPR).
L'origine di questo termine deriva da fenilchetonuria, da cui la sigla con cui è conosciuto questo disturbo (PKU). Si tratta di una malattia geneticamente trasmessa che è caratterizzata dall'alterazione di alcuni componenti chimici del corpo, che può portare a disabilità intellettuali.
Le persone nate prive dell'enzima fenilalanina idrossilasi (gli enzimi permettono di attivare alcune reazioni chimiche nel corpo), che risiede nel fegato, non possono sintetizzare la fenilalanina dal cibo e questa comincia ad accumularsi eccessivamente nel corpo. Va notato che le persone senza la malattia possono sintetizzare la fenilalanina e convertirla in L-DOPA, un neurotrasmettitore essenziale per il normale funzionamento del neurosviluppo. Ci sono sette aminoacidi essenziali per le persone (leucina, triptofano, valina, isoleucina, tirosina, metionina e fenilalanina) che sono ottenuti dalla sintesi di alcuni componenti presenti in diversi alimenti.
Anche se è una malattia meno conosciuta, si sa che solo negli Stati Uniti, circa 25.000 bambini all'anno nascono con la PKU, e anche se la sua origine precisa è sconosciuta, si sa che è più comune negli individui i cui antenati erano nord europei o indiani nativi americani.
Questa malattia genetica, che viene trasmessa da padre a figlio, ha avuto in Ivar Asbjørn Følling uno dei suoi principali divulgatori. L'esperto medico norvegese fu responsabile della descrizione della malattia nel 1934, prima di qualsiasi altro specialista.
Følling ha notato che gli enzimi menzionati sopra idrossilano la fenilalanina, in una reazione che permette la produzione di tirosina. Ciò significa che se il corpo manca di uno di questi enzimi, soffrirà di un eccesso di fenilalanina nel sangue (poiché non sarà convertita in tirosina), con conseguenti altri disturbi.
L'accumulo di fenilalanina e di altre sostanze (come il fenilpiruvato) causa danni al cervello e al sistema nervoso centrale. Chi soffre di PKU può sperimentare di tutto, dal deterioramento mentale alle convulsioni e agli spasmi, ai tremori e alle eruzioni cutanee.
I sintomi dei bambini con PKU includono capelli, occhi e pelle di colore più chiaro rispetto ai loro fratelli (la fenilalanina è una delle principali responsabili della produzione di melanina, che permette la colorazione della pelle e dei capelli), capacità intellettuali ritardate, una testa sottodimensionata, movimenti a scatti e iperattività, convulsioni e tremori e una postura insolita della mano. Bisogna aggiungere che coloro che non sono trattati o che evitano gli alimenti contenenti fenilalanina cominciano ad espellere uno strano odore (paragonabile a quello dei topi o della muffa) attraverso la loro pelle, le urine e l'alito; l'odore è dovuto all'accumulo di fenilalanina nel corpo.
Diagnosi e trattamento
Grazie ai test e alle diagnosi che possono rilevare la fenilchetonuria nei bambini appena nati, è possibile trattare la condizione aiutando questi individui a crescere e svilupparsi in modo sano. Il trattamento per la PKU di solito comprende una dieta che esclude gli alimenti con elevate quantità di fenilalanina, come le uova o il latte.
Per individuare la PKU, si eseguono semplici esami del sangue (in alcuni paesi questo test è uno dei test obbligatori per i neonati), e se positivo, si eseguono ulteriori test per confermare la diagnosi e procedere al trattamento.
È importante sapere che questa malattia è perfettamente curabile e che basta una serie di misure preventive per garantire una crescita sana ai bambini con PKU. Il trattamento comprende una dieta a basso contenuto di fenilalanina che richiede una stretta supervisione professionale, che, se seguita correttamente, garantirà una vita adulta molto più sana dal punto di vista fisico e mentale che se non viene eseguita in modo intransigente.
Alimenti come il latte, le uova e tutti quelli che contengono aspartame (come il dolcificante artificiale Nutrasweet) contengono fenilalanina e sono i primi da evitare quando si segue una dieta di questo tipo.
Ci sono alternative appositamente sviluppate per le persone che soffrono di questa malattia che permettono loro di condurre una vita sana senza dover fare a meno di alimenti che sono essenziali per la crescita, come il latte. Il latte in polvere per neonati Lofenalac, per esempio, è usato come fonte di proteine per individui con fenilchetonuria, che è fatto con un basso contenuto di fenilalanina e bilanciato in termini di quantità di altri componenti essenziali per la crescita, come gli aminoacidi.
Inoltre, è di vitale importanza che i genitori che hanno figli con questa malattia abbiano una chiara consapevolezza delle conseguenze che potrebbero colpirli se la malattia non viene trattata, o se la dieta raccomandata dagli specialisti non viene seguita correttamente, compreso un ritardo mentale cronico che si presenterà nel primo anno di vita e che sarà irrevocabile. Allo stesso modo, se i genitori non sanno se possono portare il gene di questa malattia nei loro geni, è essenziale che quando pensano di concepire una nuova vita facciano fare le rispettive analisi enzimatiche, per essere preparati, nel caso i loro figli possano essere portatori di questo disturbo.
Infine, le donne che soffrono di fenilchetonuria devono seguire una dieta rigorosa, non solo prima della gravidanza, ma anche durante la gravidanza e l'allattamento, anche se il bambino nasce senza il gene difettoso, perché l'accumulo di questa sostanza nel corpo della madre potrebbe avere conseguenze sul corpo del bambino.
